πίσω
πίσω
COPYRIGHT © by Michaelidou Maria
Dr. Maria Michailidou
SYSTEMIC SCLEROSIS (SCLERODERMIA)
General Considerations
Systemic sclerosis (scleroderma) is a chronic multisystem disease that belongs to the family of systemic autoimmune disorders. The word scleroderma literally means "hard skin" and describes the most dramatic clinical feature of the disease—namely, skin fibrosis. Scleroderma effects approximately 20 new patients per million per year and has an estimated prevalence of approximately 250 patients per million in the United States. As with many other autoimmune disorders, scleroderma is approximately 4–5 times more common in women than men. The average age at the time of diagnosis is approximately 50 years.
The prevalence and manifestations of scleroderma vary among racial and ethnic groups. For example, the disease is approximately 100 times more common among the Choctaw Native Americans in Oklahoma, in whom the disease is characterized by diffuse skin disease and pulmonary fibrosis. Milder, "limited" disease is more common among white women, and African American patients are more likely to have severe lung disease. The finding of various subtypes of scleroderma among different ethnic or racial groups, the presence of familial clustering, and the appearance of specific autoantibodies that are associated with specific HLA types suggest genetic influences on disease expression. Certain environmental factors are also thought to play etiologic roles. For example, characteristic antibodies and scleroderma disease manifestations can develop in coal miners exposed to high levels of silica.
Clinical Findings in scleroderma
Scleroderma is a rare disorder but is characterized by symptoms that occur frequently
in the general population, such as Raynaud phenomenon, gastroesophageal reflux, fatigue,
and musculoskeletal pain. Therefore, it is important for primary care practitioners
to be aware of scleroderma because early intervention can reduce morbidity and potentially
prevent life-
The diagnostic criteria for scleroderma include either thickened (sclerodermatous) skin changes proximal to the metacarpophalangeal joints or at least two of the following:
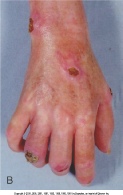
1. Sclerodactyly. 2. Digital pitting (residual loss of tissue on the finger pads). 3. Bibasilar pulmonary fibrosis.
A diagnosis of limited scleroderma can be made if the patient has three of the five features of the CREST (calcinosis, Raynaud phenomenon, esophageal dysmotility, sclerodactyly, telangiectasias) syndrome.
Patients with definite Raynaud phenomenon who have abnormal nailfold capillary loops and the presence of autoantibodies known to be associated with scleroderma can be considered to have early scleroderma or a mild expression of the disease.
Although skin changes are usually the major diagnostic clues, scleroderma is a systemic disease that most commonly targets the peripheral circulation, muscles, joints, gastrointestinal tract, lung, heart, and kidney. Symptoms encountered in the early presentation of scleroderma include musculoskeletal discomfort, fatigue, weight loss, and heartburn associated with gastroesophageal reflux disease (GERD). When these symptoms are accompanied by the new onset of cold sensitivity or Raynaud phenomenon, then scleroderma should be considered and further diagnostic investigation is warranted.
Symptoms and Signs in scleroderma
Skin
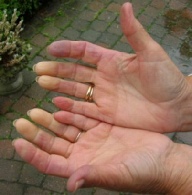
Thickening of the skin is the most easily recognizable manifestation of scleroderma but is not prominent in all patients. Patients with scleroderma are typically classified based on the amount and location of skin involvement. Patients with "limited" disease have skin changes on the face and distal to the knees and elbows. One form of limited scleroderma, the CREST syndrome, typically only involves the skin of the fingers (sclerodactyly) distal to the metacarpophalangeal joints. In contrast, diffuse scleroderma refers to the group of patients with proximal extremity or truncal skin involvement . The amount of skin thickening can be quantified by performing a "skin score," in which the skin is pinched between the examiner's thumbs in 17 specified areas of the patient's body, scoring the thickness of the skin from 0 (normal) to 3 (very thick). The skin score provides a systematic approach to longitudinal disease evaluations. Moreover, epidemiologic studies indicate that higher skin scores correlate with greater degrees of internal organ involvement and worse overall prognosis.
Vascular Disease
Involvement of the vasculature is ubiquitous among patients with scleroderma. A diffuse vasculopathy of peripheral arteries is manifested pathologically by intimal proliferation, activation of the arterial smooth muscle, and narrowing or occlusion of the vessel lumen. Critical ischemia occurs in the tissues when vasoconstriction occludes these diseased vessels. Evidence suggests that this vascular disease is fundamental to organ damage and subsequent malfunction of the heart (cardiomyopathy), lung (pulmonary hypertension), kidney (scleroderma renal crisis [SRC]), and other organs in scleroderma
.
Lung Involvement
Two main forms of lung disease occur in patients with scleroderma: inflammatory alveolitis
leading to interstitial fibrosis and pulmonary hypertension. These two processes
can occur independently or concomitantly. Lung involvement usually presents as dyspnea
on exertion but can be asymptomatic early in the course of the disease.Pulmonary
vascular disease with or without fibrosis can lead to pulmonary hypertension and
ultimately right heart failure. Isolated pulmonary hypertension occurs more commonly
in patients with limited scleroderma who have a long duration of disease. Pulmonary
hypertension complicates the disease course in approximately 10% of patients with
the CREST syndrome. Typically, patients with isolated pulmonary hypertension seek
medical care complaining of dyspnea on exertion; however, signs of progressive, life-
Renal Involvement
Clinically important kidney disease occurs in only a minority of patients, but when
it develops, renal disease poses a major threat to life. SRC develops in approximately
10% of patients. It is characterized by the sudden onset of malignant hypertension
that, if untreated, can lead rapidly to renal failure and death. Prior to the discovery
that angiotensin-
Cardiac Involvement
Cardiac involvement in scleroderma can frequently be demonstrated by objective testing
(eg, echocardiography, thallium scan, or electrocardiogram), but is usually subclinical.
Cardiopulmonary morbidity is seen primarily in the late stages of diffuse scleroderma.
Ischemia-
Musculoskeletal Involvement
Musculoskeletal symptoms range from mild arthralgias to frank nonerosive arthritis with synovitis resembling rheumatoid arthritis. The sclerosis of the skin of the fingers or limbs is often associated with contractures of the joints. Deeper tissue fibrosis can also involve the fascia and underlying muscle. If areas around the tendons are involved, active and passive range of motion of the joints are limited and painful. The physician can appreciate this on examination by feeling a "tendon friction rub" when placing the hand over the tendons as the patient flexes and extends the joint. Tendon friction rubs are found most commonly around the ankles, wrists, or knees.
Muscle weakness is a common complaint with a variety of causes including pain, prolonged
muscle disuse, malnutrition, and a slowly progressive fibrosis of striated muscle.
A true inflammatory myopathy is seen in a small subset of patients. Patients with
"overlap" phenotypes who have scleroderma features (eg, Raynaud phenomenon, interstitial
lung disease, and sclerodactyly) and a true inflammatory polyarthritis or polymyositis
may be categorized as having mixed connective tissue disease. Patients with mixed
connective tissue disease have high-
Other Symptoms scleroderma
Sicca complex (dry eyes and dry mouth) are common in patients with scleroderma but are usually not as severe as in patients with primary Sjögren syndrome. Pain is very common and usually is associated with digital ulcers, fibrosis of tendons, joint contractures, or musculoskeletal disease. Rarely, neuropathic pain is present secondary to carpal tunnel syndrome or trigeminal neuralgia.
Depression is frequent among patients with scleroderma but does not correlate directly with disease severity. Depression probably reflects other factors such as degree of pain, personality traits, and social support systems.
Erectile dysfunction is very common among men with scleroderma and is often not detected or properly managed. Fortunately, erectile dysfunction in scleroderma patients can respond to conventional therapy such as sildenafil. Sexual dysfunction among women is also common; symptoms include vaginal dryness, dyspareunia, and vaginal tightness.
Laboratory Findings
There is no single laboratory study or test that confirms the diagnosis of scleroderma. The diagnosis is made by obtaining a careful history and performing a physical examination. However, autoantibodies are found in nearly every patient with scleroderma (sensitivity > 95%). ANAs are the most frequently detected, but they are not specific for scleroderma